



















Regulatory Rapporteur
June 2024 | Volume 21 | No.6
Abstract
The need for early collaboration between regulators and European Union’s (EU) Health Technology Assessment (HTA) bodies when developing ‘fit-for-purpose’ evidence generation plans has long been acknowledged, and the ‘Health Technology Assessment (EU) 2021/2282’ (EU-HTAR) Regulation will be the first permanent collaboration between the European Medicines Agency (EMA) and HTA bodies in Europe. The framework for the EU-HTAR will include workstreams on joint clinical assessments (JCAs), joint scientific consultations (JSCs), the identification of emerging health technologies and the development of common HTA procedures and methodologies with a key role for the EMA planned in each case.
Indeed, the first implementing act of the EU-HTAR on JCAs carved out a key role for the EMA and proposes timelines in close synergy with the marketing authorisation (MA) process. Other implementing acts are set to be published throughout 2024 and will further define the role of the EMA through exchange of information, management of conflict of interest (draft published) and the process for JSCs, although this role is yet to be defined at the time of writing.
Introduction
The need for early collaboration between regulators and the EU’s Health Technology Assessment (HTA) bodies when developing ‘fit-for-purpose’ evidence generation plans has long been acknowledged, however collaboration is not always common practice. In the face of increasingly innovative products and parallel cost containment pressures, close interaction between these stakeholders is critical for ensuring access and availability of new medicines.
A Regulation on HTAs, the ‘Health Technology Assessment (EU) 2021/2282’ has been developed to harmonise the approach to HTAs across the EU, however the Regulation will also address the need for collaboration through a permanent partnership between the EMA and national HTA bodies.[1]
Overview of the EU-HTAR
The EU-HTAR entered into force in January 2022 and applies as of January 2025.[1] The Regulation will have a phased roll out but will eventually become mandatory for all medicines with a new active substance that receive a MA in the EU under the foreseen centralised procedure. The roll out will be:
- January 2025: oncology and advanced therapy medicinal products (AMTPs)
- January 2028: orphan medicinal products
- January 2030: all medicinal products[1]
The framework for the EU-HTAR will include workstreams on joint clinical assessments JCAs, JSCs, the identification of emerging health technologies and the development of common HTA procedures and methodologies.[1] Stakeholder engagement is a key objective for the EU-HTAR, and the EMA is expected to have a key role in each of these workstreams although only the role regarding JCAs has been defined at the time of writing.
A Member State (MS) Coordination Group on Health Technology Assessment (HTACG), including representatives of HTA bodies from MS, has been established to coordinate the roll out of the regulation and six ‘implementing acts’ are being developed to operationalise the different workstreams.[2] At the time of writing, the implementing act on the process for JCAs for medicinal products has been adopted and a draft of the conflict of interest management implementing act has also been published. The other acts on JCAs for medical devices (Q4), JSCs for medicinal products (Q3) and medical devices (Q4) and exchange of information with the EMA (Q3) are expected through 2024.[2]
Development of the EU-HTAR: a history of EMA collaboration
The development of the EU-HTAR is the culmination of many years of collaborative efforts between the national HTA bodies within the EU. The EU-HTAR replaces the current system of national assessments as well as the EU-funded European Network for Health Technology Assessment (EUnetHTA 21).[3] EUnetHTA 21 was the final phase in a series of initiatives developed to pave the way for a future EU HTA system, originally established as the ‘EUnetHTA project’ in 2006.[4]
The first EMA-EUnetHTA work plan was established in 2012, and the EMA has played a key role in the EUnetHTA 21, most notably in the development of a parallel JSC process of which, seven were conducted as part of the EUnetHTA 21.[3] JSCs allow manufacturers to seek advice from both regulators and HTA bodies on their clinical study designs and could be increasingly important in the context of major changes to the EU’s overarching legislation governing the pharmaceutical industry.[5] JSCs will be important for manufacturers to discuss key changes that affect both regulatory and HTA decision making including orphan designation, accelerated approval pathways and comparative clinical trials.[5] However, the implementing act on the process for JSCs has not been published at the time of writing.
Alongside JSCs, the EMA and the EUnetHTA 21 have collaborated on a number of additional projects to prepare MS for entry into the EU-HTAR. These have included:
- Product-specific discussions on the evidence generation needs for AMTPs in oncology.
- HTA involvement in the EMA initiative to establish an EU network of experts on patient-reported outcomes (PROs).
- Delivering trainings for patients and healthcare professionals to facilitate their participation in regulatory and HTA processes.
- Sharing of horizon scanning activities.
- Optimisation of regulatory assessment reports for use in the context of HTAs.
- Developing guidelines for collection of real-world evidence (RWE).[3]
Key principle of the EU-HTAR – Joint Clinical Assessments (JCAs)
The EU-HTAR aims to streamline the current EU HTA system by increasing efficiencies and removing duplication. Under the current system, national HTA bodies complete individual HTA assessments to evaluate the relative effectiveness and added value of a product in order to inform pricing and reimbursement decisions. However, HTA assessments have become increasingly complex with HTA bodies using widely different methods and criteria to inform their decisions.
Under the new Regulation, the evaluation of clinical domains of a HTA or relative effectiveness will be conducted at the EU level, providing MS with a single harmonised JCA.[1] The JCA process aims to reduce this complexity, taking place in parallel to the centralised procedure. In this procedure, the European Commission ultimately provides a single MA to all MS based on the recommendations following assessment by the EMA’s Committee for Medicinal products for Human Use (CHMP) of a medicinal product’s safety, quality and efficacy.[6]
However, unlike a centralised MA, the JCA will be non-binding and will not equate to a pricing or reimbursement decision. National HTA bodies will still be required to assess the non-clinical aspects (economic, social and ethical aspects) of a product in order to determine the added value.1 As such, pricing and reimbursement decisions will remain the sovereignty of individual MS.
Joint Clinical Assessments – a key role for the EMA
The first implementing act, titled ‘Commission Implementing Regulation (EU) 2024/1381’, was adopted on 23 May 2024 and outlines the process for conducting JCAs for medicinal products, managed by the HTACG, with a key role for the EMA alongside manufacturers.[7]
The process for JCAs will take place in parallel to the EMA centralised procedure MA process with clearly defined responsibilities for both the manufacturer and the EMA (see Figure 1). Where applicable, the JCA will be initiated at the same time as the MA submission and should be terminated in the event of a negative opinion or withdrawal from the EMA process.[7]
Figure 1: Timeline of the EMA and JCA collaboration[7]
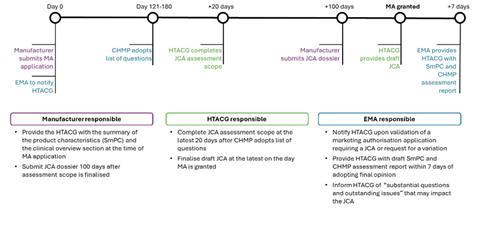
(purple =manufacturer responsibilities; blue=EMA responsibilities; green=HTACG responsibilities)
The first implementing act for JCAs outlines a key role for the EMA, however it should be noted that the implementing act has been met with criticism from some manufacturers on the long-time allowed for scoping versus the short timelines for manufacturers to prepare a JCA dossier. The timelines in the adopted implementing act were revised, after public consultation, but concerns remain. This concern may intensify as the current draft of the new general pharmaceutical legislation proposes to shorten the centralised procedure assessment timeline from 210 to 180 days which would further expedite the timelines for preparing a JCA dossier given that the timelines are dependent on the CHMP process.[5] The practical considerations of this implementing act are still yet to be understood.
Next steps
The EU-HTAR will be the first permanent collaboration between the EMA and HTA bodies in Europe and is the endpoint of many years of collaborative efforts and initiatives. As expected, the first implementing act carved out a key role for the EMA with timelines in designed to be aligned with the centralised procedure MA process. The next implementing acts will be published throughout 2024 and will further define the role of the EMA through exchange of information, management of conflict of interest in EU HTA (draft published) and the process for JSCs.
Importantly, the new regulation signals that manufacturers should no longer think of regulatory and HTA decision making as separate processes. Evidence generation plans should be developed through close interactions between both decision makers in order to improve the possibility of approval and hence access to novel therapies for patients.
References
[1] European Commission. Regulation (EU) 2021/2282 of the European Parliament and of the Council of 15 December 2021 on Health Technology Assessment and Amending Directive 2011/24/EU.
[2] European Commission. Implementation Rolling Plan 2023-2024.
[3] European Medicines Agency and EUnetHTA (2023) Report on the Implementation of the EMA-EUnetHTA 21 Work Plan 2021 - 2023
[4] EUnetHTA Secretariat. (2009) EUnetHTA Project. Overview of results, years 2006-2008
[5] European Commission. Regulation of the European Parliament and of the Council Laying down Union Procedures for the Authorisation and Supervision of Medicinal Products for Human Use and Establishing Rules Governing the European Medicines Agency, Amending Regulation (EC) No 1394/2007 and Regulation (EU) No 536/2014 and Repealing Regulation (EC) No 726/2004, Regulation (EC) No 141/2000 and Regulation (EC) No 1901/2006.
[6] European Medicines Agency. (2019) From Laboratory to Patient: The Journey of a Medicine Assessed by EMA
[7] European Commission. Commission Implementing Regulation (EU) 2024/1381 of 23 May 2024 laying down, pursuant to Regulation (EU) 2021/2282 on health technology assessment, procedural rules for the interaction during, exchange of information on, and participation in, the preparation and update of joint clinical assessments of medicinal products for human use at Union level, as well as templates for those joint clinical assessments.





