


















Digital health technologies have revolutionised the global healthcare system. This continuing professional development supplement provides an overview of the qualification frameworks for digital health technologies, which are used as drug development tools in the EU and the US.

In recent years, digital health technologies have revolutionised the global healthcare system; they include a broad range of products and services such as wearable devices, telemedicine, mobile health (mHealth), and health information technology. These tools have the potential to improve the ability to accurately diagnose and treat diseases, as well as to enhance the accessibility and delivery of care. They include technologies intended for use as a medical product, in a medical product, as a companion diagnostic, or as an adjunct to other medical products (ie, devices, drugs, and biologics). In addition, they may also be used to develop or study another medical product. Using data from digital health technologies in the benefit–risk evaluation of medical products can also raise questions about how these data, and the endpoints they support, compare with conventional data capture. This article provides an overview of the EU and US regulatory frameworks for digital health technologies used as drug development tools.
The EU regulatory landscape
As digital health technologies span a broad range of products and applications, developers may need to follow regulations that apply to medicinal products, medical devices, in vitro diagnostics, or a combination of these regulations. In addition, the EU’s General Data Protection Regulation governs the handling of personal data relating to individuals in the EU.[1] Developers that collect, use, and process patient data captured via digital health technologies will need to comply with this regulation. The European Medicines Agency (EMA) is primarily responsible for the review of data generated by digital health technologies that support the development, use, or monitoring (pre- or post-marketing authorisation) of medicinal products. Specifically, a key consideration for the agency is the expected role of the technology in the development, evaluation, or use of a medicinal product.2 Digital health technologies that meet the definition of medical devices or in vitro diagnostics are regulated by the national competent authorities of EU member states. Two new regulations on medical devices and in vitro diagnostics entered into force in May 2017 and are progressively replacing the existing directives. Refer to the CPD covering the fundamentals of the European regulatory framework for medical devices[3] for further details.
Qualification of digital health technologies to support development and marketing The EMA qualification of novel methodologies is a voluntary scientific process to establish the acceptability of a new digital health technology for the development of medicinal products.[4] Digital technologies within the scope of the qualification programme include digital endpoints that are derived from or include digital measures, digital biomarkers, and electronic clinical outcomes assessments.[2] Definitions of the digital biomarker and electronic clinical outcome assessments are provided in Table 1.
The qualification process results in either a qualification opinion or qualification advice:[4]
- The Committee for Medicinal Products for Human Use (CHMP) qualification opinion is a public document detailing the acceptability of a specific use of a new methodology in a research and development context (eg, use in pre-clinical or clinical studies). The applicant submits protocols, study reports, and supportive data to establish using the defined novel methodology for a specific purpose. One relevant example is the EMA qualification opinion on eSource Direct Data Capture in clinical trials.[5]
- The CHMP qualification advice is a confidential document that provides advice on future protocols and methods for further scientific development towards qualification. In this case, the applicant submits draft protocols and development plans for future studies (intended to establish using the defined novel methodology for a specific purpose), a scientific rationale, and any available supportive data.
| EMA [2] | FDA[16] | ||
|---|---|---|---|
| Electronic clinical outcome assessment | Digital biomarker | Clinical outcome assessment | Biomarker |
| Quantifiable measure used as a measure of how patients feel, function or survive that is derived from a digital measure. Clinical meaning is established de novo. | Objective, quantifiable measure of physiology and/or behaviour used as an indicator of biological, pathological process or response to an exposure or an intervention that is derived from a digital measure. Clinical meaning is established by a reliable relationship to an existing, validated endpoint. | Measurement of a patient’s symptoms, overall mental state, or the effects of a disease or condition on how the patient functions (eg, patient-reported outcome, clinician-reported outcome, performance outcome) [FD&C Act, section 507 (e)(3)] | Characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathological responses or biological responses to a therapeutic intervention, and includes a surrogate endpoint (FD&C Act, section 507 (e)(1)]. |
Overview of the qualification procedure
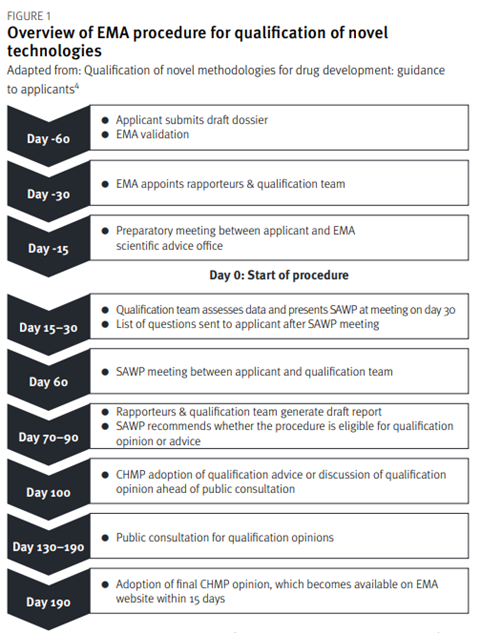
The qualification of novel methodologies for drug development is summarised in Figure 1. The applicant’s submission of a request is followed by a 60-day presubmission phase. Before submitting the request, applicants are encouraged to review the EMA’s essential considerations for a successful qualification of novel methodologies[6]. During the pre-submission phase, the dossier is validated, the qualification team is appointed, and a preparatory meeting is held. Following the successful outcome of pre-submission, the procedure then starts on Day 0 and the EMA appointed coordinators begin their review of the dossier.
The qualification team assesses the data and presents its initial assessment at a meeting with the Scientific Advice Working Party (SAWP).[4]The qualification team experts assess the dossier and produce a list of questions for the applicant. A discussion then takes place with the applicant and the qualification team at a meeting. After this meeting, the SAWP generates a draft report and recommends whether the procedure is eligible for a qualification opinion or qualification advice.[4] The CHMP then either discusses and adopts the qualification advice or discusses the qualification opinion. For qualification advice, the applicant receives an advice letter providing input on the studies required to generate data for a future qualification opinion. For a qualification opinion, the draft opinion is published for a six-week public consultation. Based on the feedback from the scientific community, discussions, and the data submitted, the procedure may conclude with a final adoption of the qualification opinion by the CHMP. The procedure’s target duration is 100 days for the qualification advice and 190 days for the qualification.
The US regulatory landscape
In the US, data stemming from digital health technologies that support drug or biologic development are reviewed by the Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER). Digital health technologies classified as medical devices are regulated by the Center for Devices and Radiological Health (CDRH). Refer to our previous CPD article covering US medical device regulations[7] for further information. In September 2020, the FDA launched the Digital Health Center of Excellence (DHCoE) to support the agency’s goal of advancing digital health technologies.[8] The center of excellence is a network of experts in multiple areas of digital health technology who provide centralised expertise on digital health technologies and related policy for the FDA. [9]The DHCoE supports the review process for digital health technologies across the FDA but is not responsible for marketing authorisation decisions.[10] For the public, the DHCoE also has links to educational materials, guidance documents, and ways to submit questions on interpreting those guidances.
The center of excellence also is continuing to expand and develop resources for external stakeholders including international regulatory agencies, healthcare providers, researchers, industry, and the public.[11] The FDA does not have a single dedicated pathway for drug development tools such as digital health technologies. Digital health technologies that generate data intended for use as a clinical outcome or a biomarker can be assessed through an investigational new drug (IND) application, the scientific community consensus pathway, or the Drug Development Tool (DDT) Qualification Programs.[12] Through a direct submission to FDA as part of an IND application, a sponsor can bring forward technological ideas and negotiate with subject matter experts in the FDA’s clinical division. The negotiations may cover the utility of the tool and how it might be used in a clinical trial setting. The scientific community consensus pathway typically goes through publication in scientific journals or consensus statements put forth by professional societies.
This approach can be useful for hypothesis generation, but often does not make primary data as readily available to the FDA as the IND pathway does. Consequently, the tools that come through this pathway do not tend to be as ‘regulatory ready’. One example of this lengthy process is the monitoring of HbA1c to determine how well diabetes is controlled. First proposed in 1976,[13] it was not until 2008 that the FDA published a guidance on using HbA1c as a well-validated surrogate endpoint for certain short-term and long-term complications of diabetes.[14] The DDT Qualification Program provides for the review and acceptance of a method, material, or measure that aids in drug or biologic product development and regulatory assessment based on a qualification package. Qualified DDTs are then available for use in any development programme and regulatory application within the context of use (COU) defined in the qualification package. The COU describes the circumstances under which the DDT is to be used in drug/biologic product development and regulatory review.15 The COU serves the same purpose in both the EU and the US – defining how the digital health technology will be used – although each agency includes different elements in their respective COU definitions.
| Clinical Outcome Assessment (COA) Qualification | Biomarker qualification | ||
|---|---|---|---|
| Letter of intent | Letter of Intent to Propose COA Qualification[17] | Biomarker Qualification Letter of Intent (LOI) Content Elements[20] | |
| Qualification plan | COA Qualification Plan[18] | Biomarker Qualification Plan: Outline of Suggested Content Elements[21] | |
| Full qualification package | COA Full Qualification Package[19] | Biomarker Full Qualification Package[22] | |
| Letter of intent |
|
||
| Qualification plan | |||
| Full qualification package | |||
Overview of the DDT Qualification Program
The FDA has three DDT qualification programmes for biomarkers, clinical outcome assessments, and animal models.[15] Table 1 has definitions of the biomarker and clinical outcome assessments. The animal model qualification programme applies only to animal models intended for use in animal efficacy studies for drugs developed under regulations commonly known as the Animal Rule. The qualification procedure includes three stages, namely, letter of intent (LOI), Qualification Plan (QP), and Full Qualification Package (FQP). Table 2 provides key documents for each stage of the biomarker and clinical outcome assessment programmes. For comparison, EMA key documents are also highlighted in Table 2. The LOI submission initiates the process after which the DDT committee, comprising CDER and CBER subject matter experts, scientists, and senior level medical officers, determine whether to accept the DDT into the relevant qualification programme. If accepted, a LOI Determination Letter is issued with recommendations, considerations, and requests to the applicant on the next step, the QP. In addition to addressing recommendations from the LOI, the QP should describe relevant data, knowledge gaps, and proposed data collection and analysis plans. If the QP is acceptable, the FDA issues a Determination Letter that includes requests for data and recommendations about data needs for the FQP. The FQP is the comprehensive, final stage and includes study protocols and reports, statistical analysis plans, summary and subject-level data related to the DDT and its COU. The FQP review considers whether the evidentiary standards for the DTT in its proposed COU have been met. If successful, this review concludes with a Determination Letter indicating the qualified status of the DDT. If a not-qualified determination is made, the applicant can address the FDA input and resubmit a modified FQP or submit a new LOI. There are no preset meetings in any of the qualification process. The applicant can request interactions with the agency before or during the qualification process by contacting the appropriate qualification program and following its process for arranging meetings. Transparency provisions apply to the qualification submissions and information is posted to the qualification programmes websites.[15]
Conclusion
Both the EMA and the FDA have established voluntary processes to qualify digital health technologies that support the development of medicinal products. The EMA and FDA qualifications are multi-stage processes that are supported by progressively more robust data packages. Given the nature of the qualification process, applicants benefit from collaboration and frequent engagement with multiple stakeholders throughout the development process, including early interactions with regulators.
For the case study please click here: www.regulatoryrapporteur.org/cpd-supplements/oct-21-cpd-case-study-qualification-of-stride-velocity-endpoint-in-duchenne-muscular-dystrophy/35.article
To register your CPD, please use our CPD tool: https://www.topra.org/TOPRA_Member/Professional_Development/CPD_and_lifelong_learning/CPD_tool/TOPRA/TOPRA_Member/CPD/MyCPD.aspx
[1] . Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC (General Data Protection Regulation) (OJ L 119, 4.5.2016, p. 1)
[2] Questions and answers: Qualification of digital technology-based methodologies to support approval of medicinal products. June 2020. EMA. Available at: https://www.ema.europa.eu/en/documents/ other/questions-answers-qualification-digital-technology-basedmethodologies-support-approval-medicinal_en.pdf
[3] Purnama A, Drago D. Fundamentals of the European Medical Devices Regulatory Framework. Regulatory Rapporteur. October 2019. Available at https://www.topra.org/TOPRA/TOPRA_MEMBER/PDFs/ TOPRA-RR-OCT19-CPD-Supplement.pdf
[4] Qualification of novel methodologies for drug development: guidance to applicants. October 2020. EMA. Available at: https://www. ema.europa.eu/en/documents/regulatory-procedural-guideline/ qualification-novel-methodologies-drug-development-guidanceapplicants_en.pdf
[5] Qualification opinion on eSource Direct Data Capture (DDC). July 2019. EMA. Available at: https://www.ema.europa.eu/en/documents/ regulatory-procedural-guideline/qualification-opinion-esource-directdata-capture-ddc_en.pdf
[6] Essential Considerations for a successful qualification of novel methodologies. December 2017. EMA. Available at: https://www.ema. europa.eu/en/documents/other/essential-considerations-successfulqualification-novel-methodologies_en.pdf
[7] Purnama A, Drago D. FDA regulatory pathways for medical devices. Regulatory Rapporteur. May 2019. Available at: https://www.topra.org/ topra/topra_member/pdfs/CPD-May-2019-Medical-Devices-and-FDA.pdf
[8] FDA Launches the Digital Health Center of Excellence. 22 September 2020. FDA. Available at: https://www.fda.gov/news-events/pressannouncements/fda-launches-digital-health-center-excellence (accessed 12 January 2021).
[9] Network of Digital Health Experts. October 2020. FDA. Available at: https://www.fda.gov/medical-devices/digital-health-centerexcellence/network-digital-health-experts (accessed 15 January 2021).
[10] About the Digital Health Center of Excellence. 22 September 2020. FDA. Available at: https://www.fda.gov/medical-devices/digitalhealth-center-excellence/about-digital-health-center-excellence (accessed 15 January 2021).
[11] Ask a Question About Digital Health Regulatory Policies. November 2019. FDA. Available at: https://www.fda.gov/medical-devices/ digital-health-center-excellence/ask-question-about-digital-healthregulatory-policies (accessed 26 January 2021).
[12] Leptak C. Drug Development Tools (DDTs) Regulatory Perspective. Presented at FDA Public Meeting on 29 March 2019. Available at: https://www.fda.gov/media/125220/download (accessed 26 January 2021).
[13] FDA Case Study Biomarker Qualification. March 2017. FDA. Available at: https://www.fda.gov/media/104459/download
[14] Guidance for Industry – Diabetes Mellitus: Evaluation Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes. December 2008. FDA. Available at: https://www.fda.gov/ media/71297/download
[15] Qualification Process of Drug Development Tools Guidance for Industry. November 2020. FDA. Available at: https://www.fda. gov/regulatory-information/search-fda-guidance-documents/ qualification-process-drug-development-tools-guidance-industryand-fda-staff
[16] Biomarker Qualification: evidentiary framework guidance for industry. December 2018. FDA. Available at: https://www.fda.gov/ regulatory-information/search-fda-guidance-documents/biomarkerqualification-evidentiary-framework
[17] Letter of Intent to Propose COA Qualification. June 2020. FDA. Available at: https://www.fda.gov/media/117148/download
[18] COA Qualification Plan. June 2020. FDA. Available at: https://www.fda. gov/media/123245/download
[19] COA Full Qualification Package. June 2020. FDA. Available at: https:// www.fda.gov/media/128005/download
[20] Biomarker Qualification Letter of Intent (LOI) Content Elements. January 2019. FDA. Available at: https://www.fda.gov/media/120058/ download
[21] Biomarker Qualification Plan: Outline of Suggested Content Elements. January 2019. FDA. Available at: https://www.fda.gov/media/120059/ download
[22] Biomarker Full Qualification Package. June 2019. FDA. Available at: https://www.fda.gov/drugs/biomarker-qualification-program/ biomarker-full-qualification-package















