

















The regulations, developed as a result of the 1976 Medical Device Amendments to the Food, Drug, and Cosmetic Act of 1938, share a common goal with the pharmaceutical regulations: they both strive to ensure that new medical treatments reach the public as quickly as possible while protecting patients and ensuring that the new treatments have a positive benefit–risk balance. However, they approach this goal in different ways. This continuing professional development supplement explains the fundamentals of the FDA regulatory pathways for medical device manufacturers that wish to bring their products to the US market.
In 1976, amendments to the Federal Food Drug and Cosmetics (FD&C) Act expanded the US FDA’s responsibility to also oversee medical devices, in addition to its drug role,[1] under the Center for Devices and Radiological Health (CDRH). There are many similarities between the medical device and the pharmaceutical regulations. However, the pace of innovation in these two fields is different. Whereas a new drug approval takes an average of 10 to 15 years, moving a new medical device from concept to market takes an average of three 3 to seven 7 years.[2]
According to the FD&C Act, a device is “an instrument, apparatus, implement, machine, implant or an in vitro reagent or other similar or related article, including a component part, or accessory” that meets three conditions:[3]

1) Recognised in the official National Formulary or the US Pharmacopoeia;
2) Intended for use in the diagnosis of disease or other conditions, or the cure, mitigation, treatment, or prevention of disease, or;
3) Intended to affect the structure or function of the body of humans, and which does not achieve its primary intended purposes through chemical action or by being metabolised.[4]
The range of objects that falls under the FDA definition of medical devices is broad, for example, tongue depressors, stethoscopes, laboratory equipment, surgical instruments, pacemakers and ventilators. Some products that contain biological material are inert (eg, acellular dermatologic fillers) and can also be considered devices.[5] ,[6]
Bringing a device to market
The development of an entirely new device typically begins with a concept by a physician or a bioengineer for a solution to a medical problem. A preliminary prototype of the device is built and simultaneously a patent process is initiated. Preliminary bench testing is then followed by animal testing, and the device enters a cycle of testing and redesign.
Although portrayed as a compartmentalised process with distinct phases, such as preclinical and clinical, steps in device development overlap and portions may need to be repeated as testing and user experience are incorporated into product modifications and the device moves closer to its marketed form. There are at least three key steps that developers should follow to bring their device to the US market:[7]
Step 1: Classify the device;
Step 2: Select the appropriate regulatory pathway; and
Step 3: Register the establishment and list the device
Step 1: Classify the device
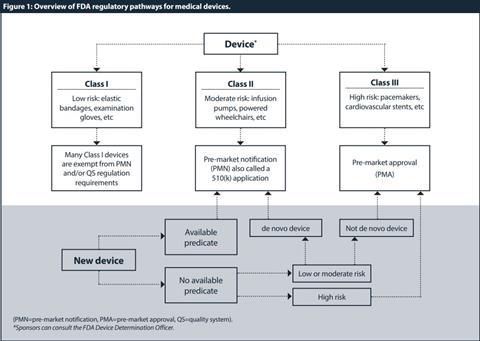
The first step, after determining that the product is a device, is to classify the device. Because medical devices vary widely in their complexity and benefits or risks, they do not require the same degree of regulation. Thus, the FD&C Act established the riskbased device classification system for medical devices. Each device is assigned to a regulatory class based on the level of control necessary so that there is a reasonable assurance of its safety and effectiveness. Device classification depends on intended use and indications for use. All devices are classified into three groups by the FDA:[8] [9] [10] Class I or “low risk”; Class II or “medium risk”; and Class III or “high risk” devices.
Class I devices have the least regulatory requirements. Under current law, Class I devices are defined as those for which general controls “are sufficient to provide reasonable assurance of the safety and effectiveness of the device”.[11] Many Class I devices are exempt from the pre-market notification and/or the quality system (QS) regulation requirements. [12] [13]
Class II devices are defined as those “which cannot be classified as class I because the general controls by themselves are insufficient to provide reasonable assurance of safety and effectiveness of the device”.[14] Class II devices can only be marketed after providing the FDA with a “pre-market notification”, also called a “510(k)” submission.[15] Only a few Class II products require studies in humans to support claims of performance or safety. For the majority of Class II products, requirements can be satisfied by bench and animal testing.
Class III devices include devices which are life-supporting or life-sustaining and present a high or potentially unreasonable risk of illness or injury to a patient. Unless the manufacturer obtains a reclassification, all new devices that are not Class I or II are automatically designated as Class III.[16] Before a Class III device is marketed it must be approved by the FDA. This is different than for Class II devices, which are “cleared” by the agency.
Once a device is classified, an appropriate regulatory pathway needs to be conducted. The summary of regulatory pathways for medical devices by the FDA is described in Figure 1.
Step 2: Select the appropriate regulatory pathway
Pathway 1—Pre-market approval (PMA)
A PMA is a stringent type of marketing application required by the agency for new or high-risk devices. The PMA approval is based on a determination by the FDA that the application contains sufficient evidence to provide reasonable assurance that the device is safe and effective for its intended use(s). [17] PMAs generally require some clinical data before an approval decision. All clinical evaluations of investigational devices (unless exempt) must have an investigational device exemption (IDE) before the clinical study is initiated.[18] An IDE allows an unapproved device to be used in a clinical study to collect the data required to support a PMA submission.
The content of the PMA includes: (1) summaries of nonclinical and clinical data; (2) a device description; (3) indications for use; (4) a description of the foreign and US marketing history; (5) the proposed labelling; and (6) a description of the manufacturing process. Approval is based not only on the strength of the scientific data but also on the inspection of the manufacturing facility to ensure that the facility and the manufacturing process comply with the quality system (QS) regulation.[19] The PMA process can be described in 4 steps as depicted in Table 1.
Pathway 2—Pre-marketing notification (PMN), also known as 510(K) application
In general, a 510(k) submission is required for a moderate-risk medical device that is not exempt from pre-market review. The standard for clearance of a traditional 510(k) is substantial equivalence with a predicate device, which can be either a previously cleared Class I or II device that does not require a PMA, or a pre-amendment Class III for which the agency has not issued regulations requiring a PMA. There are three types of 510(k) submissions for pre-market clearance: traditional, special, or abbreviated.
A traditional 510(k) application includes the device’s name, description, intended use, proposed label, as well as a comparison with a predicate device, and the device’s advertisement and directions for use,[20] supported by preclinical studies. The term substantial equivalence, in many cases, means simply that the device performs in a similar fashion to the predicate under a similar set of circumstances. Many Class II devices that are cleared via a 510(k) submission do not have to demonstrate safety and effectiveness through clinical studies with human subjects.
A special 510(k) application is appropriate when the manufacturer is planning device modifications to its own legally marketed device (predicate device). Such modifications may not affect the intended use or the fundamental scientific technology of the device.
An abbreviated 510(k) is appropriate when the manufacturer is planning to rely solely on the use of guidance documents, special controls, and recognised standards.
Some novel devices without a predicate have another alternative pathway available called de novo. Under the FD&C Act, novel devices lacking a legally marketed predicate are automatically designated Class III. In 1997 the act was amended to allow the FDA to establish a new, expedited mechanism for reclassifying these devices based on risk. The amendment resulted in a reduction of the regulatory burden on manufacturers. The de novo application, although requiring more data than a traditional 510(k), often requires less information than a PMA application.[21] Devices approved as de novo can serve as predicates for other devices.
Pathway 3—The Humanitarian Device Exemption
The Safe Medical Devices Act of 1990 authorised the Humanitarian Device Exemption (HDE)[22] to encourage the development of devices that aid in the treatment and diagnosis of diseases or conditions affecting less than 4,000 individuals in the United States per year. To encourage manufacturers to develop devices for these small markets, the HDE application is similar to a PMA but it is exempt from the effectiveness requirements. The device sponsor is only required to demonstrate that there is a probable benefit to health and that the probable benefit outweighs the risk of injury or illness caused by the device. In other words, the HDE requires demonstration of device-related safety, but not device efficacy.[23]
However, there are some important restrictions such as, for example, a 4,000-unit limit per year on the number of devices shipped. In addition, the use of an HDE device requires approval by an institutional review board (IRB) at the institution where the device is to be used.
| Table 1: Application timeline for 510(k) and PMA. | |
|---|---|
| Pre-market notification (PMN) – 510(k) | Pre-market approval (PMA) |
|
Day-1 FDA receives 510(k) submission |
Day-1 FDA receives PMA submission |
|
Day-7 FDA sends Acknowledgement Letter or Hold Letter if unresolved issues with User Fee and/or eCopy |
Day-45 FDA verifies if the application is administratively complete. Or else, the application will be returned |
|
Day-15 FDA conducts Acceptance Review or FDA informs sponsor if 510(k) is accepted for Substantive Review or placed on RTA Hold |
Day-120 FDA completes the Initial Review and determines if an advisory committee meeting is necessary |
|
Day-60 FDA conducts Substantive Review of FDA communicates via a Substantive interaction to inform the sponsor that the FDA will either proceed with Interactive Review or that the 510(k) will be placed on hold and Additional Information is required. |
Day-180 (+) FDA regulations allows 180 days to review and make a determination. However, the total review time can be much longer. There are four options for the final deliberation: a) approval order b) approvable letter c) not approvable letter d) order denying approval |
|
Day-90 FDA sends final MDUFA Decision on 510(k) |
|
|
Day-100 If MDUFA Decision is not reached by Day 100, FDA provides Missed MDUFA Decision Communication that identifies outstanding review issues |
|
| (RTA=refuse to accept, MDUFA=medical device user fee amendments) | |
Step 3: Register the establishment and list the device
The FDA requires all medical device manufacturers to register their facilities and list their devices with the agency. Manufacturers and initial distributors of medical devices must register their establishments with the FDA. All establishment registrations must be submitted electronically unless a waiver has been granted by the FDA. All registration information must be verified annually. In addition to registration, foreign manufacturers must also designate a US agent.[24]
Post-market regulations and processes
Once their product is approved or cleared for marketing, manufacturers of medical devices must comply with various regulations on labelling and advertising, manufacturing, and post-marketing surveillance. The iterative nature of medical device development adds a layer of complication. It is challenging to create postmarket requirements for a product that may be replaced by the next-generation product before the start of, for example, a post-market surveillance study.
The current US medical device post-market surveillance system depends primarily on the following sources for detecting potential problems with medical devices:[25]
• Medical device reporting (MDR) The FDA annually receives several hundred-thousand reports of suspected medical device-related malfunctions, serious injuries, and deaths
• Medical Product Safety Network (MedSun) The FDA receives about 5,000 higher quality reports each year on device use and adverse outcomes from a network of 280 US hospitals
• Post-approval studies Such studies may be ordered by the FDA as a condition of approval for a PMA device
• Post-market surveillance studies The FDA may order a manufacturer of a Class II or Class III device to conduct post-market surveillance studies
• FDA discretionary studies The FDA conducts its own research to monitor device performance, investigate adverse event signals and characterise device-associated benefits and risks to patient subpopulations.
Conclusion
Although the FDA’s process to bring new medical devices to the US market can be daunting, the agency has implemented activities to increase the transparency and predictability of the process. Numerous guidance documents and the FDA policies and procedures are available on the FDA’s website. Medical device developers and manufacturers are also ‘encouraged to take advantage of the opportunities available for meetings with FDA officials.
[1] Federal Food Drug and Cosmetics Act P.L. 75-717, 1938, x505(c) and (d).
[2] KM Fargen, D Frei, D Fiorella et al. ‘The FDA approval process for medical devices: an inherently flawed system or a valuable pathway for innovation?’ J Neurointerv Surg 2013;5:269–75.
[3] Federal Food Drug and Cosmetics Act 21 U.S.C. 321(h).
[4] US FDA. Classification of products as drugs and devices and additional product classification issues. Available at: http://www.fda.gov/RegulatoryInformation/Guidances/ ucm258946.htm (accessed 15 November 2018).
[5] US FDA. Soft tissue fillers (dermal fillers). Available at: https://www.fda.gov/MedicalDevices/ ProductsandMedicalProcedures/CosmeticDevices/ ucm619837.htm (accessed 15 November 2018).
[6] US FDA. Device — Not a device. Available at: http://www. fda.gov/MedicalDevices/DeviceRegulationandGuidance/ Overview/ClassifyYourDevice/ucm051521.htm (accessed 15 November 2018).
[7] AV Kaplan, DS Baim, JJ Smith et al. ‘Medical device development: from prototype to regulatory approval’. Circulation 2004;109:3068–72.
[8] US FDA. CFR-Code of Federal Regulations Title 21 vol. 8. Sec 880. 5240. Medical adhesive tape and adhesive bandage. Available at: http://www.accessdata.fda.gov/ scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?FR¼880.5240 (accessed 15 November 2018).
[9] US FDA. CFR-Code of Federal Regulations Title 21 vol. 8. Sec 878.5020. Nonabsorbable polyamide surgical suture. Available at: https://www.accessdata.fda.gov/scripts/ cdrh/cfdocs/cfcfr/cfrsearch.cfm (accessed 15 November 2018).
[10] US FDA. CFR-Code of Federal Regulations Title 21 vol. 8 Sec 870.3610 Implantable pacemaker pulse generator. Available at: http://www.accessdata.fda.gov/scripts/ cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr¼870.3610 (accessed 15 November 2018).
[11] 21 C.F.R. 860.3.
[12] FFDCA §513(a)(1)(A).
[13] QS regulation is found in 21 C.F.R. §820.
[14] FFDCA §513(a)(1)(B).
[15] US FDA. Overview of medical device regulation, Medical Device Classification, Class I/II Exemptions. Available at: http://www.fda.gov/MedicalDevices/ DeviceRegulationandGuidance/Overview/ ClassifyYourDevice/ucm051549.htm (accessed 15 November 2018).
[16] FFDCA §513(a)(1)(C).
[17] US FDA. Pre-market approval (PMA). Available at: http://www.fda.gov/MedicalDevices/ DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/ default.htm (accessed 15 November 2018).
[18] US FDA. Requests for feedback on medical device submission: The presubmission program and meetings with Food and Drugs Administration staff. Available at: http://www.fda.gov/downloads/MedicalDevices/ DeviceRegulationandGuidance/GuidanceDocuments/UCM311176.pdf (accessed 15 November 2018).
[19] US FDA. Medical devices, PMA application contents. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/ HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/ ucm050289.htm (accessed 15 November 2018).
[20] US FDA. Medical devices, how to prepare a traditional 510(k), content and format of a traditional 510(k), 07/01/2015. Available at: http://www.fda.gov/ MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/ PremarketSubmissions/PremarketNotification510k/ucm134572.htm#link_4 (accessed 15 November 2018).
[21] US FDA. Evaluation of automatic Class III designation (De Novo) summaries. Available at: https://www.fda.gov/AboutFDA/CentersOffices/ OfficeofMedicalProductsandTobacco/CDRH/CDRHTransparency/ ucm232269.htm (accessed 15 November 2018).
[22] US FDA. Humanitarian Device Exemption. Available at: http://www.fda.gov/ MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/ PremarketSubmissions/HumanitarianDeviceExemption/default.htm (accessed 15 November 2018).
[23] C.F.R. §814.100, §814.104(b)(5), and §814.124
[24] US FDA. How to register and list. Available at: http://www.fda.gov/ MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/ RegistrationandListing/ucm053185.htm (accessed 15 November 2018).
[25] US FDA. Medical Devices, regulatory controls. 26 June 2014. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/ Overview/GeneralandSpecialControls/default.htm#gen (accessed 15 November 2018).











