

















This continuing professional development article aims to provide some of the background and key steps and considerations when using and navigating the EU centralised procedure for a new marketing authorisation application (MAA).
Regulation 726/2004/EC[1] lays down community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishment of the European Medicines Agency (EMA). The centralised procedure (CP) makes provision for submission of a single new marketing authorisation application (MAA) to the EMA with scientific assessment being conducted by a rapporteur/co-rapporteur, and assessment of the risk management plan by the Pharmacovigilance Risk Assessment Committee (PRAC). Scientific discussion and final opinion are carried out by the Committee for Human Medicinal Products (CHMP), resulting in one EU marketing authorisation (MA) for the EU issued by the European Commission (EC).

Although access to the CP is restricted, EU CENTRALISED PROCEDURE Key steps and considerations of the EU centralised procedure one of the main objectives is to provide a common assessment procedure whereby the CHMP provides scientific opinion by consensus or majority with independent reviews (two primary assessments) by the rapporteur and co-rapporteur. This serves to facilitate a common decision-making process (EC decision) and greater transparency (through the provision of European public assessment reports [EPARs]).
The CP requires the use of one (invented) name, one marketing authorisation holder (MAH), one contact person and common product information (summary of product characteristics [SmPC], labelling and patient leaflet) in all official languages, resulting in an MA that is valid in all EU member states and the European Economic Area (EEA). It also provides for a faster Europe-wide approval for those companies wishing to commercialise in multiple markets and, by virtue of having a single licence, there are potential lifecycle management resource and cost savings. However, from a commercial viewpoint, due to differences in disease prevalence across the EU, a pan-EU marketing authorisation may not be necessary for your product and having a single MAH, tradename and pack limits marketing flexibility.
When it comes to organising your company for a CP MAA, having a sound and well considered project plan and the right resources to execute it is of vital importance to the success of your application, as is managing company expectations. In addition to assessing the product’s eligibility, a data package gap analysis is an invaluable exercise to conduct early in product development, and possibly as part of the decision-making process in whether to select the CP. This task serves both to aid generation of the correct data during development but also in the submission planning stage to ensure the best possible chance of a successful validation and likelihood of a positive opinion. This is best done with a cross-functional team (clinical, preclinical and quality), which should convene to conduct a gap analysis on the data package. The team should review available guidance and legislation; review EPARs for authorised medicinal products with similar indication(s) or issues, eg, comparator(s)/standard of care (SOC) used, efficacy endpoints, safety profile, specification limits, stability data, toxicity profile etc; list all known and potential issues with data package (missing data, unfavourable results, etc) and rank in terms of risk to approval (critical, major, minor).
The output of this review should enable you to develop a risk mitigation strategy so you can determine which issues can or should be: resolved pre-submission; resolved with a postapproval commitment; justified based on the overall benefit–risk profile and unmet medical need; or determine which issues should be discussed with the (co)rapporteur and/or EMA.
| PRAC |
|---|
|
• Responsible for all aspects of the risk management of medicines • Detection, assessment, minimisation and communication on risk related to medicinal products • Periodic safety update reports and signal assessment • Design and evaluation of post-authorisation safety studies • Assessments of urgent/non-urgent Union procedure triggered due to safety concerns (safety referral procedures). • Regulatory oversight of risk management plan (RMP) and assessment of outcome of risk minimisation measures in RMP • Advice to CHMP on selected post-authorisation procedures. |
| CAT |
|
• Preparation of draft opinion on each advanced therapy medicinal product (ATMP) application before the CHMP adopts a final opinion • Upon request, provide opinion on any scientific matter relating to ATMPs • Participate in procedures for ATMP classification and certification • Contribute to other procedures and activities as needed (eg, scientific advice for ATMPs). |
| COMP |
|
• Deliver opinions on orphan drug designation applications • Provision of advice to EU Commission on policy on orphan medicines and assisting in drafting detailed guidelines • Assisting in international cooperation • Link with patients associations. |
| PDCO |
|
• Assessment of content of paediatric investigation plans (PIPs) and applications for full or partial waivers and deferrals • Assessment of data generated in accordance with agreed PIPs • Adopting opinions on quality, safety and efficacy of a medicine for use in the paediatric population at request of CHMP or national competent authority • Provision of advice to member states on content and format of data to be collected through surveys on the uses of medicines in children • Advising and supporting the development of the European Network of Paediatric Research at the EMA • Providing advice on questions on paediatric medicines • Establishing and updating an inventory of paediatric medicine needs. |
Organisations and committees involved in the CP
The EMA is an EU agency which was founded in 1995. Under the supervision of a management board, the scientific secretariat of approximately 800 full-time staff is responsible for coordinating the existing scientific resources put at its disposal by member states for the evaluation, supervision and pharmacovigilance of medicinal products (as per Article 55 of Reg. (EC) No 726/2004). The EMA has seven scientific committees (see Table 1):
- CHMP Committee for Human Medicinal Products
- PRAC Pharmacovigilance Risk Assessment Committee
- CAT Committee for Advanced Therapies
- COMP Committee for Orphan Medicinal Products
- PDCO Paediatric Committee
- CVMP Committee for Veterinary Medicinal Products
- HMPC Committee for Herbal Medicinal Products.
The CHMP is composed of a chairperson, elected by serving CHMP members; one member (and an alternate) nominated by each of the EU member states; one observer, non-voting member (and an alternate) nominated by each of the EEA states Iceland and Norway; up to five co-opted members, chosen among experts nominated by member states or the EMA to gain additional expertise in a particular scientific area. Therefore, there are up to 33 voting members and opinion voting is by simple majority (17 votes required for a positive opinion, with divergent opinions recorded).
There are also a number of working parties, including biologics (BWP), patients’ and consumers’ (PCWP), safety (SWP), quality (QWP), pharmacogenomics (PgWP), vaccines (VWP) and biostatistics (GTWP), which conduct the scientific work of the agency. Scientific advisory groups (SAGs) can be convened for cardiovascular, antiinfectives, neurology, diabetes/endocrinology, HIV/viral diseases, vaccines, oncology, psychiatry and diagnostics topics. There is also an ad hoc expert group.
Agendas, minutes and meeting highlights from all scientific committees are published on the EMA website. Via these scientific committees and working parties, the EMA is responsible for conducting the initial assessment of EUwide MAAs and any modifications (variations) to an existing MA. It delivers opinions to the European Commission and to the World Health Organization (WHO), provides scientific advice and protocol assistance to companies, prepares scientific guidelines and regulatory guidance and interacts with regulators (eg, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use process and the US FDA).
The EMA has published important information to help users of the CP prepare for the expected consequences of Brexit, including their obligations relating to establishment within the EEA. Further information is available in the UK’s withdrawal from the EU section on the EMA website.[2]
CP scope
Mandatory scope (under Article 3(1) of Reg (EC) No 726/2004) covers those new products for which the CP must be used to ensure that important medicines are able to be made available across the whole of the community. The categories of products that fall under the mandatory scope are: recombinant DNA technology; controlled gene expression; hybridoma and monoclonal antibody methods; similar biological (“biosimilar”) medicinal products; ATMPs (gene therapy medicinal products; somatic cell therapy medicinal products; tissue engineered products); orphan medicinal products; and new active substances for AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune diseases, other immune dysfunctions, viral diseases.
For products outside the mandatory scope, provision is made within the legislation for other product categories to make use of the CP. In the case of an optional scope application – Article 3(2)(b) – you will need to prepare a suitable justification either for demonstration of significant innovation or provide an argument that your company’s product would, if approved, be in the interest of patients at the community level. This case might apply, for example, in the event you are registering a new indication for an established active ingredient. Other examples include paediatric use marketing authorisation (PUMA) applications, or applications including a paediatric indication included in a paediatric investigation plan (PIP).
The CP is not exclusively limited to new active substances and makes provision for the approval of generic medicines (including biosimilars) based upon previously approved innovator products with centralised authorisations. However, a centralised application for a generic (or hybrid) of a product approved nationally or via the mutual recognition procedure or decentralised procedure is possible as long as a suitable justification can be given that it would represent a significant therapeutic, scientific or technical innovation, or that its authorisation would be in the interest of patients at a community level. The CP assessment process therefore begins 18 to seven months before day 0 with submission of eligibility request.
CP assessment
A number of activities take place approximately seven months before day 0, including submitting a notification of intent to submit an application, appointment of rapporteurs, as well as a potential pre-submission meeting. To assist the agency in ensuring resource is available and assessment of the application can be factored into ongoing workloads, the EMA advises applicants to consider the date of submission carefully, referring to the published submission dates and guidance.[3] Upon receipt of the intention to submit an application, the CHMP and PRAC will appoint (co)-rapporteurs to conduct the scientific assessment of the dossier. For ATMPs, (co)-rapporteurs are also appointed from members of the CAT who will lead the assessment. When the (co)-rapporteurs have been appointed it can often be of great value to organise a face-to-face meeting in order to introduce the dossier and to seek clarification on any specific aspects of the dossier that might be an issue during review.
Although not mandatory, the pre-submission meeting will normally take place six to seven months before submission of your MAA and provides the best opportunity for applicants to obtain procedural and regulatory advice from the agency. The aim of these meetings is to allow the applicant to discuss any legal basis, regulatory and procedural topics with the goal of ensuring a smooth validation phase and, along with a sound knowledge and reference to relevant guidance, provision of data to maximise the chances of a positive assessment of the application.
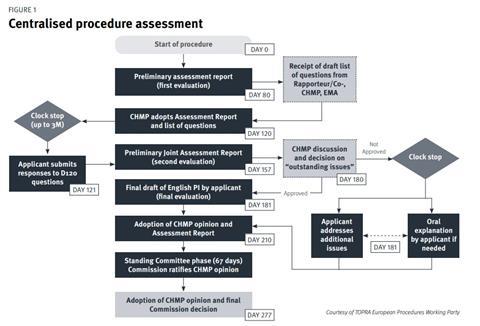
The standard CP (Figure 1) comprises two main assessment phases which take place over a 210-day period (excluding clock-stops where the applicant answers assessment questions).[4] The primary evaluation takes place from day 1 to day 120, at which point the applicant will need to respond to the list of questions posed by the CHMP (you have up to three months to respond). Following on from this is the secondary evaluation from day 120 to day 180 wherein a second round of questions follows (list of outstanding issues). Following the month clock-stop for the company to respond to day 180 questions, the final evaluation and CHMP opinion is issued at day 210, which is followed by a further phase of 67 days – the decisionmaking phase at the end of which the European Commission issues its decision on the grant of the MA.
Although these are the main steps, there are a few possible additional steps that might occur such as routine or triggered requests for good manufacturing practice (GMP), good laboratory practice (GLP), good clinical practice (GCP) inspections, consultation with scientific advisory groups (SAGs) or ad hoc expert groups, and other committees and working parties. A number of committees are therefore involved during the CP. There is also the possibility that the company may be asked to give an Oral Explanation at the CHMP.
EMA product team and applicant interaction
An EMA product team will be set up for each medicinal product submitted through the CP. [5]This team is responsible for providing support to the evaluation activities of the EMA scientific committees. The team is established during the presubmission phase of the initial MAA and is in place throughout post-authorisation.5 The composition of the team may change over time depending on the complexity of the product and procedure, as well as the type of issues raised during the product’s lifecycle.5 This team oversees all elements of product knowledge. The applicant will be notified when both the EMA procedure manager (PM) and an EMA product lead (EPL) have been appointed.[6]
The PM is the applicant’s primary contact during the course of all evaluation procedures. PMs can provide regulatory procedural guidance, ensure adherence to procedural guidelines and timelines and offer regulatory scientific support in simpler procedures. They also maintain process performance metrics.
The EPL is responsible for the EMA product team and is ultimately accountable for overall product knowledge. They provide clinical and regulatory science input, support consolidation of a committee position and facilitate crosscommittee discussions. Depending on the activity, during the procedure you will need to make a note of the correct person to contact. The applicant should contact the PM for all questions regarding the evaluation procedure, including: requests for guidance in the pre-submission phase, such as the pre-submission meeting; any type of procedural questions during the evaluation, such as availability of assessment reports and opinion documents; and discussion on timetables, including requests for extension of clockstops, etc. At certain milestones the EPL and the applicant will be in direct contact to facilitate the discussion on the scientific evaluation, eg, clarification meetings; immediate feedback from committee plenary discussions; oral explanation expectations; discussion of required postauthorisation measures; and late-stage revisions of the product information before adoption of the final opinion.
The first port of call during evaluation of the MAA will be the PM. Any questions concerning validation of the MAA, once submitted, will be dealt with by an assigned validation officer. Occasionally other members from the EMA product team may contact you directly to facilitate discussion on specific aspects (eg, quality, risk management, mock-up review). If you are in direct contact with the EPL (or another member of the EMA product team) the PM should always be copied in on any correspondence.
[1] European Medicines Agency. Reg. (EC) No 726/2004. Available at: https://ec.europa. eu/health/sites/health/files/files/eudralex/ vol-1/dir_2001_83_consol_2012/dir_2001_83_ cons_2012_en.pdf (accessed 22 March 2019).
[2] European Medicines Agency. Brexit – UK’s withdrawal from the EU. Available at: https://eur-lex.europa.eu/content/ news/Brexit-UK-withdrawal-from-the-eu. html (accessed 22 March 2019).
[3] European Medicines Agency. General guidance on application and evaluation. Available at: https://www.ema.europa. eu/en/human-regulatory/marketingauthorisation/marketing-authorisationguidance-documents#Application and evaluation (accessed 22 March 2019).
[4] European Medicines Agency. Steps involved in obtaining an EU marketing authorisation. Available at: www.ema. europa.eu/en/human-regulatory/marketingauthorisation (accessed 22 March 2019).
[5] European Medicines Agency. EMA presentation: Overview of recent changes in the centralised procedure. Presented by Michael Berntgen, Head of Scientific and Regulatory Management Department, at 2nd Industry stakeholder platform, 9 November 2015. Available at: https://www.ema.europa.eu/en/documents/ presentation/presentation-overview-recentchanges-centralised-procedure-michaelberntgen_en.pdf (accessed 22 March 2019).
[6] European Medicines Agency. Presubmission guidance: questions and answers. Available at: https://www.ema.europa. eu/en/human-regulatory/marketingauthorisation/pre-authorisationguidance (accessed 22 March 2019).











