
















This continuing professional development (CPD) supplement focuses on the regulatory complexities and challenges associated with biosimilar products and their development. Although biosimilar products have been registered and approved for use in the EU for more than a decade, there is increasing speculation and excitement on the potential for biosimilars with increasingly complex structures, eg, multi-subunit, extensively post-translationally modified, and lipid-containing products.
The European Medicines Agency (EMA) describes biosimilar medicines as “a biological medicine that is similar to another biological medicine that has already been authorised for use”.[1] Similarity is evaluated and established against other, EU-registered and established biopharmaceutical products. These are referred to as reference medicinal products. The reference product must have been authorised for at least ten years before a biosimilar being made available, enabling a robust and extensive clinical and adverse events profile to be established.

By their very nature, biopharmaceuticals are intrinsically variable. Therefore, biosimilars cannot be referred to as generics, a term applied to pharmaceutical products denoting “sameness” between products. There are, nevertheless, a number of other terms that are more widely accepted descriptors of biosimilars, including: follow-on biologic (FOB), follow-on protein (FOP), and subsequent entry biologic (SEB) that are favoured by other regulatory bodies.
Given the complexities of these biological molecules, it is important to highlight and address these complexities and their impact on manufacturers and patient groups. In particular, this article considers some of the anticipated limitations when moving beyond the currently registered products into more complex molecules. Although no complex biosimilars are yet approved, they are in development and will need to be understood in the context of what the current frameworks require.
Background and history
The complex nature and batch-tobatch variability of biological quality attributes within acceptable ranges is very well known. Manufacturers and regulators appreciate and accept that the manufacturing process, controls, limits and specifications will vary due to harnessing living cells to manufacture a target molecules, the associated intrinsic variability of the manufacturing process and have expectations set accordingly. Given their unique properties for treatment of serious diseases, many biological products have attracted a premium price in terms of reimbursement for patient care costs, consequently making them attractive to be copied (following patent expiry). Their high value has also made these products less available to a wider population of patients and furthermore is another driver of the biosimilars market. Consequently, there are a disproportionately high number of manufacturers in south Asia and the Far East who are singularly driven to produce biosimilars. During the late 1980s and 1990s, a range of biological products based around growth hormones and monoclonal antibodies (mAbs) were developed and successfully registered. Given their novel, proprietary and often complex manufacturing processes and applications, the intellectual property (IP) around many of these products was protected by various patents.
However, due to this relatively new pharmaceutical landscape, the biosimilars industry is confronted with a constantly evolving regulatory environment. The first biosimilars guidelines were issued by the EMA in 2005;[2] additional EMA guidance has since been released and some of the initial guidelines have even been revised.1 Thereafter, other highly regulated countries followed (Japan in 2009, Canada in 2010) and in 2012, the US FDA released its first draft biosimilar guidelines[3] [4] [5] Thus, in this constantly evolving regulatory environment, the biosimilars industry needs to ensure that its ongoing and planned developments comply with the criteria in the regulations and guidelines.
Changes in the regulatory landscape included the development of guidance for “wellcharacterised” (later renamed “well-specified”) biologics in the US, “comparability protocols, and “equivalence protocols” in the EU. Although these changes in the regulatory requirements were intended primarily to support and facilitate changes to biologics manufacturing processes, they triggered the evolution of the concept of the biochemical bridge, whereby a comprehensive analytical (biochemical and biophysical) comparative testing programme for structural and functional characteristics could form the basis of the justification for demonstration of similarity.
The biochemical bridge easily lent itself to the analysis of other “similar” biologics and to start to define differences and correlate to physiological and clinical effects. Clinical differences should be avoided, however. Some differences are expected, and hence dose alignment and management in reference to biosimilar switching might be required. More recently, manufacturers have been encouraged and guided to assess the “totality of evidence” when making evaluations on the similarity of biological products. The FDA has published all available data for biosimilars assessment since around 2012. The concept of “totality of evidence” seeks to encompass and assess a wide range of parameters and attributes that may not have traditionally been included as part of product release. For example, the methods of manufacture can plan a significant role in the quality of a biological product, thus the evaluation of manufacturing data and controls can play a part in product assessment. These concepts have formed a pivotal part of the biosimilars registration framework, and continue to underpin the development and registration of these products.
“First wave” biosimilars
Biological products are generally more complex than pharmaceutical preparations that result from chemical synthesis. The complexity of biological products compared with small molecules results from a number of factors unique to biological molecules, including:
•Molecular mass (typically 10kDa or more)
• Composition (protein, carbohydrate, lipid, nucleic acid, cell debris)
• Higher order structure (rendering biological function)
• Complex manufacturing operations/ processes
• Formulation, including the use of adjuvants
Given these and many other limitations (related to IP, manufacturing complexity issues, etc), the first wave of approved biosimilars were relatively simple biological molecules with limited (often just glycosylation) post translational modifications. They were still, nevertheless, vastly more complex than small molecule pharmaceuticals. These limitations should also not be considered individually because, often, they are interwoven or interlinked, particularly for more complex biosimilars such as Remsima (infliximab, a chimeric entity). As such, progress with the development and registration of biosimilars is generally hampered by a number of aspects, including:
• Complexity regarding the innovator molecules:
- IP
- Demonstrating equivalence with intrinsically variable innovator, and showing batch-to-batch variability due to its natural structure and biological function
• Limitations placed on biosimilar interchangeability, substitution, switching, and extrapolation of indication
• Complex multi-subunit or multimodal biologics (eg, antibody-drug conjugates [ADCs], vaccines)
• Data requirements for registration (analytical and biochemical, nonclinical, clinical)
• Other “unknown” considerations, typically where the issue has not arisen or has not been identified yet for this class of molecules (keep an open mind when looking at the apparent totality of data).
Data requirements
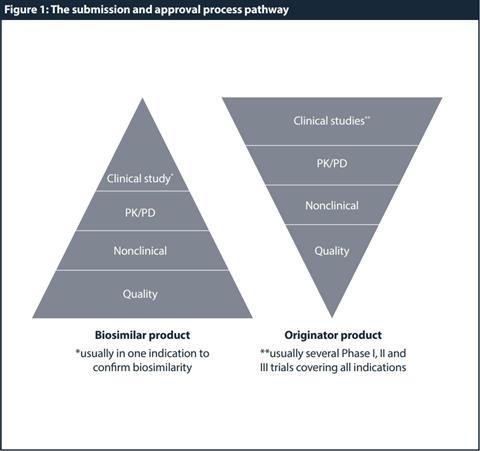
Based on these special properties and criteria, a generic development and approval (ie, demonstration of bioequivalence with the reference product by bioavailability studies) is therefore not sufficient. Instead, full quality development complemented by a comprehensive comparison of the physicochemical and biological parameters with the reference product is required. At the physicochemical level and due to the comparative testing and characterisation, the biosimilar developer is required to provide sufficient quality data to demonstrate parity with the originator. The extent and nature of the nonclinical and clinical data package should be tailored to detect potential differences between the biosimilar and the reference product. Hence, demonstration of patient benefit per se is not the only scope, but to establish similarity to the reference product and consequently, to allow partial reliance on efficacy and safety data collected for the reference product. Figure 1 shows a high-level comparison of the requirements for a biosimilar versus originator/reference product development. Understanding these regulatory requirements and trends in biosimilar development is crucial for a successful registration.
Additionally, the data required during the review and approval of biosimilar products will vary considerably on the type of biosimilar product. Although these data will be assessed holistically during the review process, it is helpful to ensure they are determined and reported in an easy-to-understand pieces. The data may therefore be classified in a number of ways, regardless of the data is reported in common technical document (CTD) module 3.2.R, which aids comprehension of the data. This may be done by detailing and analysing the constituent components of the molecule:
•Protein
• Lipid
• Carbohydrate
• Nucleic acid/others (cell debris).
By reporting the structure of the molecule:
• Primary
• Secondary
• Tertiary
• Quaternary.
By type of testing, including functional stepwise assessments:
• Analytical/biochemical/biophysical
• Biological and immunochemical
• Nonclinical
• Clinical.
Switching
The terms interchangeability, substitution, and switching generally refer to the practice of using the originator biologic and changing to an approved biosimilar, or changing from one approved biosimilar to another approved biosimilar. Extrapolation is the term used to describe the use of a biosimilar for an indication approved for the reference product, but not for the biosimilar. There is no provision for automatic extrapolation and prior approval and sound scientific justification is required before a biosimilar may be extrapolated to other indications approved for the reference product.
Following the approval of a small molecule or pharmaceutical product, being able to switch (or substitute) between pharmaceutical drug products is a well-established and extensively used phenomenon and is typically implemented at the pharmacy level. In addition to restrictions against biosimilar extrapolation, this type of switching and interchangeability requires approval at the national level. There are a number of considerations that must be taken into account as part of interchangeability, switching, and substitution.
The biosimilar approval and review process is underpinned by an evaluation against the innovator/reference product using a totality of-evidence approach. Following its approval, the biosimilar receives its own marketing authorisation (MA) number and becomes a product in its own right, with no requirement to continue to demonstrate biosimilarity post approval. Although this is perfectly sensible, the longer term acceptability of such an approach becomes more challenging in the post-approval period. Following the approval of a biosimilar (which may be viewed as a “similarity snapshot” in time), both the biosimilar and the reference product can embark on different post-approval pathways, which may result in significant differences in their composition, structure, biological effects and, possibly in the future as manufacturers wish to capitalise on returns, expanded clinical use (eg, Botox). This potential divergence could place considerable strain on the extrapolation of the clinical and chemistry, manufacturing and controls (CMC) data reviewed at the time of biosimilar approval.
There are a wide range of analytical procedures that may be applied to biosimilars and these are well documented by others. State of the art analytical methods should indeed be used for testing and characterisation. The specifications for biological molecules typically encompass two types of variability: intrinsic variability resulting from the natural variability of a class of products, and analytical test variability resulting from the assays used to test the product. This intrinsic variability can result in specifications for the target molecule (reference product and biosimilar) that may be viewed as being “wide”.
In actual fact, it is the intrinsic variability that makes biosimilarity challenging. In addition to analytical and extensive characterisation data for the bulk and finished product, biosimilar analysis profiling should also include in-process data (and specifications) and stability data (comparative, during storage, in-use, and accelerated) because this totality of evidence will be used to determine biosimilarity. For global submissions, reference product from different jurisdictions should be sourced and analysed. If the variability is wide, this may be because of low number of lots tested. However, manufacturers of biologics may use different assays for activities rather than interchangeable methods, which adds to the complexity.
Conclusion
The approval of the current of crop of biosimilar products is, without doubt, a great achievement that has enabled greater patient access to medicines with high medical need and remains a major milestone in pharmaceutical development. With these achievements come greater challenges, not only maintaining diligence and patient safety with the currently approved products, but to develop biosimilar registration packages for biological products of increasing complexity. The difficulties in surmounting the next challenges are significant, and include detailed understanding of the structure and function relationships of biological molecules that are comprised of more than one protein molecule, complexed branched sugar chains, lipid bilayer components, and the possibility of nucleic acid and cell debris (sometimes deemed to be an impurity). Qualitative or quantitative variability in any of these components may, at best, result in loss of biological function or, in worse cases, severe (potentially unknown) adverse events.
[1] European Medicines Agency. Multidisciplinary: Biosimilar. Available at: https://www.ema.europa.eu/ en/human-regulatory/research-development/scientificguidelines/multidisciplinary/multidisciplinary-biosimilar (accessed 1 January 2019).
[2] European Medicines Agency. CHMP/437/04 Rev 1. Guideline on similar biological medicinal products. October 2014. Available at: https://www.ema.europa. eu/documents/scientific-guideline/guideline-similarbiological-medicinal-products-rev1_en.pdf (accessed 8 January 2019).
[3] PMDA. PFSB/ELD Notification No_0304007. Guideline for Ensuring Quality, Safety and Efficacy of Biosimilar Products, March 2009. Available at: https://www.pmda. go.jp/files/000153851.pdf (accessed 8 January 2019)
[4] Health Canada. Fact Sheet: Biosimilars. Available at: https://www.canada.ca/en/health-canada/ services/drugs-health-products/biologicalsradiopharmaceuticals-generic-therapies/applicationssubmissions/guidance-documents/fact-sheetbiosimilars.html (accessed 1 January 2019).
[5] FDA. Biosimilars – Guidances. Available at: www.fda. gov/drugs/guidancecomplianceregulatoryinformation/ guidances/ucm290967.htm (accessed 1 January 2019).