







Regulatory Rapporteur
July/August 2024 | Volume 21 | No.7
Abstract
The legislative requirement for the conduct of environmental risk assessments (ERAs) in the EU is not new, with the original guidance having been issued 18 years ago. However, 1 September 2024 marks the day when the revised guidance on the conduct of ERAs becomes effective, having been in the making since 2016 when the concept paper on the revision of the original ERA guidance was first released. This article discusses and analyses key changes of this revised guidance.

An environmental risk assessment (ERA) is the process of evaluating the potential risks of a medical product intended for human use on the environment. The objective is to safeguard aquatic and terrestrial ecosystems, including surface water, groundwater, soil, species vulnerable to secondary poisoning, and microbial processes in sewage treatment plants (STPs). ERAs must be included in all new medicinal product marketing authorisation applications (MAAs) in Europe, regardless of their route of application and legal basis, which also means that generics are not exempt of this requirement. The revised (Rev.1) guideline was issued on 15 February 2024 and replaces the current version, which has been in effect since 2006.[1],[2] Although the revised guidance officially becomes effective from 1 September 2024, the majority of concepts introduced in the guideline have been available to applicants since 2018, when the draft version of this updated guidance was first issued for public consultation.[3] Owing to the extended review and consultation process, the principles outlined in the 2018 draft guidance have been followed and implemented in practice by many regulators and applicants ever since.
The revised guideline is comprehensive (64 pages as opposed to 12 in the initial 2006 guideline) and provides more explicit detail and explanation on how to conduct an ERA and assess any environmental concerns associated with the use of a drug substance. Additionally, the guideline delineates the process for identifying potential hazards associated with the active substance of a medicinal product. It addresses potential precautionary and risk mitigation measures and offers guidance on reporting the findings in an ERA report.
The key high-level changes introduced in the 2024 guidance are herein discussed.
General principles
The original 2006 European Medicines Agency (EMA) guidance described the assessment of potential risks to the environment of a drug substance as a stepwise, phased procedure that may be terminated when sufficient information/data was available to either indicate that the drug substance was unlikely to pose a risk to the environment or to identify and sufficiently characterise the potential risks.
Although the ERA concept per the revised 2024 guideline has remained the same and is still based on the use of the product, and the physiochemical, ecotoxicological, and fate properties of the drug substance, the revised guideline is more comprehensive and provides more explicit direction on several topics. The revised guideline retains the overarching 2006 concept of the tiered approach; however, substantial changes have been incorporated, providing a more consistent and complete approach for the evaluation of potential environmental risks associated with the use of a drug substance. In terms of the approach and required testing, the following aspects of the revised 2024 guideline are particularly noteworthy:
- Both a risk assessment (exposure calculation) and a hazard (persistent, bioaccumulative and toxic (PBT) or very persistent and very bioaccumulative (vPvB)) assessment are now required and, notably, the PBT/vPvB assessment will now be done independently of the exposure calculation (risk assessment).
- A decision tree with a question-and-answer section has been added for the Phase I risk assessment to simplify the requirements and the steps of the process.
- The majority of fate and effect studies have been moved to Phase II Tier A, with Tier B focusing on refining exposure calculations.
- Technical details for studies conducted as a part of the Phase II risk assessment have been included to ensure consistency in the approach to testing and thus the data provided, and some of the triggers for further assessment and additional studies, have been amended.
- Data should be compliant with good laboratory practice (GLP), where applicable, and preferably follow the most recent test guidelines issued by the Organisation for Economic Co-operation and Development (OECD) or comparable international validated test guidelines.
- The term ‘endocrine disruptors’ has been replaced by ‘endocrine active substances’ (EAS) to include all substances potentially affecting development and reproduction.
- A new section on secondary poisoning has been introduced to estimate the accumulation of active substances in the food chain.
- The revised guideline makes provision for the use of publicly available data. This is new and specifies that for drug substances that are already marketed, information may be available in the public domain. Specific guidance on search, use and evaluation of published data is provided.
- Principles of the 3Rs (replacement, reduction and refinement) are now embedded into the guidance; as such, drug developers are encouraged to share their data to avoid unnecessary repetition of studies with living organisms. When data is shared, a letter of access should be submitted stating that the current applicant has access to data previously generated by another marketing authorisation holder (MAH). Should there be any missing studies, it is the responsibility of the current applicant to have these studies completed.
- The revised guideline further explains the conditions that will warrant a tailored ERA based on mode of action and, as anticipated, has more focus on assessing specific endpoints, such as endocrine disrupting properties for potential EAS, antibiotics, and drug substances showing PBT/vPvB properties.
- An ERA is required for all initial MAAs and is also required for certain post-authorisation applications that may lead to increased environmental exposure (e.g. extension applications), regardless of the legal basis. Notably, generics are no longer able to apply for waivers via simple reference to the legal basis of the application.
Review of the tiered approach strategy in the revised guideline
For each ERA, both a risk assessment and a hazard (PBT/vPvB) assessment are required (see Figure 1), which are incorporated into two major phases (Phase I and Phase II).
Figure 1: Overview of the risk assessment and PBT/vPvB assessment
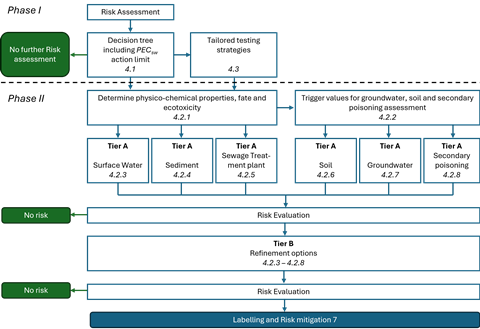
Phase I: Mandatory assessment based on environmental exposure and general characteristics of the product.
- Fundamental to Phase I is a decision tree, which identifies substances for which a Phase II ERA is required. The decision tree incorporates elements from the 2006 guideline, and is presented as effective visual guidance. The outcome is that either the risk assessment stops or a Phase II assessment is required. The rationale for not proceeding to Phase II should be included in the ERA report.
- This decision tree clarifies that a Phase II assessment is not warranted for non-natural peptides/proteins that are readily biodegradable.
- Although still part of Phase I, the PBT assessment should now be regarded as a separate and stand-alone exercise. The PBT assessment is a hazard assessment concerning the identification of intrinsic properties of an active substance that could pose harm to the environment, regardless of the levels of exposure. Therefore, this is not based on predicted environmental concentration (PEC), since cumulative effects over a longer period of time can result in uncertainty in PEC calculations.
- All drug substances (with a few exceptions) must be assessed for PBT properties, regardless of their PEC. The PBT screening (octanol/water partitioning study) occurs in Phase I, with a definitive assessment in Phase II, if the trigger value of Log Kow >4.5 is met. If a definitive PBT assessment is required, it should follow the PBT and vPvB criteria defined under the REACH regulation. To note, Section 5 of the guideline has been retitled PBT/vPvB Assessment.
- The trigger for testing has been slightly altered in the revised guideline and, to address concerns for ionisable substances, pH ranges are required in both the PBT and bioaccumulation assessments.
- The risk assessment reflects the likelihood of an effect occurring and is an evaluation of both exposure of organisms in the environment to the active substance and ecotoxicity. The revised guideline proposes the same approach and the same threshold value for the initial risk assessment (exposure calculation) for predicted environmental concentration in surface water (PECSW) (0.01 µg/L). Note, this is still restricted to the aquatic compartment. The option to refine the PECSW based on a fraction of the overall market penetration (market penetration factor: FPEN) by providing reasonably justified market penetration data (e.g. based on published epidemiological data) is retained.
Phase II: During Phase II assessment, the experimental studies are conducted to allow for a detailed fate and effects assessment. Per the revised guidance, the following studies/assessments should be considered during this phase of the risk assessment:
- determination of physicochemical properties, fate and ecotoxicity
- trigger values for soil, groundwater and secondary poisoning
- surface water
- sediment
- sewage treatment plant
- soil
- groundwater
- secondary poisoning
- tailored testing strategy for active substances with a specific mode of action
- antibacterials
- endocrine active substances (EAS).
A summary of the changes are detailed below:
- All testing has been moved to Phase II Tier A; studies that should be performed in Phase II Tier A are those on physicochemical characteristics, fate and ecotoxicity, with Tier B focusing on refining exposure calculations.
- Phase II Tier A now begins with an assessment of physicochemical properties (water solubility, dissociation in water – mandatory information) and other properties (such as UV absorption/melting point – not mandatory information). Much of this data may already be available, but testing will now need to be OECD/GLP compliant for the ERA.
- Substances entering Phase II risk assessment – the surface water, sediment and STP compartments – will now require assessment. Mandatory fate studies are included in the ERA in order to evaluate the fate and predict the environmental exposure of the drug substance.
- Studies on environmental fate properties Phase II Tier A are now limited to the ready biodegradation test and adsorption (includes assessment of three soils and two types of sludge).
- OECD test 308 (Aerobic and Anaerobic Transformation in Aquatic Sediment Systems), which is typically used to determine if a substance proceeds to sediment toxicity testing, is currently compulsory for the Phase II risk assessment per the 2006 EMA guidance. The revised guideline has replaced OECD 308 with mandatory sediment toxicity testing in Phase II (Tier A). Use of OECD 308 has been retained but will be limited to certain categories of substances, e.g. those requiring definitive PBT assessment.
- Aquatic toxicity studies and risk assessment is essentially unchanged, other than for antibiotics (OECD 201 – three species needed).
- If the drug substance meets certain trigger values, the risk assessment should be performed for soil, groundwater and/or secondary poisoning. Of note, changes have been made to the trigger for soil (terrestrial) assessment, and the exposure assessment for groundwater is now derived from the predicted no-effect concentration (PNEC)SW and includes an amended assessment factor. This will likely result in a more conservative PNEC value for most drug substances.
- The revised guidance has introduced a secondary poisoning assessment (Log Kow is ≥3 [pH 5–9]). This is primarily to assess the potential of a drug substance to accumulate through the food chain with an ultimate toxic effect on birds and mammals. It should be noted that a lack of accumulation in mammals does not exclude a potential for accumulation in fish and other aquatic species. This assessment is based on the data from the fish bioconcentration study combined with available nonclinical mammalian toxicity data.
Other notable sectional changes in the revised guideline
Section 6 ‘Search on Evaluation of Data’ is new to the revised guideline. A targeted literature review on endpoints of significance is required, even if data or a previously performed ERA is obtained from another MAH, to identify new information on ecotoxicity of the active substance. The applicant should demonstrate how the literature search was performed. The data output from the literature review should be assessed for robustness and evaluated in line with the updated guideline. Specific guidance on search, use and evaluation of published data is provided in the revised guideline.
Section 7 ‘Labelling and Risk Mitigation’ replaces ‘Precautionary and Safety Measures to be taken for Administration, Disposal and Labelling’ and includes consideration of potential precautionary and risk mitigation measures (potential labelling). When the possibility of environmental risks cannot be excluded, specific arrangements to limit the environmental impact should be made. This is discussed in Section 7, which tabulates proposed labelling aimed at minimising discharge of unused medicine into the environment. References to Summary of Product Characteristics (sections 5.3 and 6.6), Labelling (section 10) and Patient Information Leaflet (section 5) are included.
Section 9 ‘Structure of ERA Report’ replaces ‘The Environmental Risk Assessment Report’. The report should start with the new identification section clearly outlining the identification of the active substance. Summaries of all studies used should be included in the report. The full study reports and references should be provided in the annex of the ERA.
Conclusion
Although ERA per the revised guideline is still based on the use of the product, and the physiochemical, ecotoxicological and fate properties of the drug substance, the revised guideline is more comprehensive and provides more explicit direction on several topics. The September 2024 update maintains the principal of the tiered approach as per 2006; however, substantial changes have been incorporated, including the types/number of test systems recommended and additional test systems/assays for areas not, or inadequately, addressed previously.
The steps of Phase I are explicitly outlined in a decision tree, with the outcome being that either the risk assessment stops or a Phase II assessment is required. The key criterion for entering the Phase II assessment is a PECSW of ≥0.01 μg/L; however, this does not apply to EAS. Where the evidence shows that endocrine adverse effects could occur at levels below 0.01 g/L, the active substance should be further EAS assessed.
Phase II consists of two tiers: Tier A requires studies on the physicochemical properties, environmental fate and ecotoxicological effects of the active substance. It also details the trigger values that necessitate risk assessments for soil, groundwater and secondary poisoning. The PEC is then compared with the PNEC and, where risks are identified, a Tier B assessment is warranted, which includes PEC refinement options.
Overall, the revised guideline provides a more consistent and complete approach for the evaluation of potential environmental risks associated with the use of the drug substance. ERA is mandatory for an MAA as well as for some post-approval procedures. The revised guideline ensures that the ERA approach results in equal requirements for all applicants; as such, the biggest impact may be realised by producers of generic drugs, since additional testing may be necessary for some older drug substances/generics where insufficient data are available.
However, there are now possibilities for better use of data in the public domain to make a scientifically sound assessment of the environmental risk, with a special interest to avoid unnecessary repetition of animal studies (e.g. fish), and applicants are encouraged to share data in the interest of 3Rs (reduced animal testing). It addresses potential precautionary and risk mitigation measures and offers guidance on reporting the findings in an ERA report.
Acknowledgements: Thanks to Candice Kaisermann.
References
[1] Committee for Medicinal Products for Human Use (CHMP). (2006) Guideline on the Environmental Risk Assessment of Medicinal Products for Human Use (EMEA/CHMP/SWP/4447/00 corr 2, June 2006).
[2] Committee for Medicinal Products for Human Use (CHMP). (2016) Questions and answers on ‘Guideline on the Environmental Risk Assessment of Medicinal Products for Human Use’ (EMA/CHMP/SWP/44609/2010 Rev. 1, 26 May 2016).
[3] Committee for Medicinal Products for Human Use (CHMP). (2016) Concept paper on the revision of the ‘Guideline on the Environmental Risk Assessment of Medicinal Products for Human Use’ (EMEA/CHMP/SWP/4447/00 corr 2)